
Key Learning Objectives
- Discover why “salvaging” niacinamide is essential for healthy NAD+ levels.
- Find out how NAD+ is made from niacinamide (NAM).
- Learn why higher doses of NAM might inhibit sirtuins, but moderate doses don’t.
- Understand how the amount of NAD+ precursors taken affects methylation.
The Salvage Pathway: Introduction
The salvage pathway is used to produce NAD+ from nicotinamide molecules. Whether the source of the nicotinamide is vitamin B3 (as niacinamide), newer nicotinamides (e.g., nicotinamide riboside [NR], nicotinamide mononucleotide [NMN]), or molecules in food that get broken down during digestion into nicotinamide, the salvage pathway turns them into NAD+ in our tissues.
We can make the NAD+ molecule starting from three different pathways. These are (1) synthesizing it from one of the salvage pathway substrates mentioned above (i.e., NAM, NR, NMN), (2) producing it from niacin (nicotinic acid; NA) by the Preiss-Handler pathway, or (3) building the niacinamide molecule from scratch starting from L-tryptophan (Trp) using the de novo synthesis pathway. But, no matter which of these ways is used to make NAD+ the first time, the salvage pathway is the way we remake NAD+ after it gets used for cell signaling purposes (i.e., NAD+ consumption uses).
Think of the salvage pathway as doing two things. It can make NAD+ from several niacin equivalents (e.g., NAM, NR, NMN). More importantly, it allows cells to remake NAD+ over and over.
NAD+ molecules are broken apart or “consumed” in our cells every day. When an NAD+ molecule is consumed, niacinamide (NAM) is left over. To support a healthy NAD+ pool, this leftover NAM is recycled into NAD+ as needed. Cells and tissues consume far more NAD+ than we make from vitamin B3 equivalents in the diet or would get from even high doses in a supplement. Recycling the leftover NAM using the salvage pathway is what allows them to keep pace.

Figure 1. NAD+ Substates and Uses
NAD+ Consumption and Recycling Leftover NAM
Whether the niacinamide (NAM) molecule (see figure 2) comes from foods, supplements, or is split off NAD+, the same enzymes salvage it to turn it into NAD+. We wrote about what NAD+ does in a previous article. One of these jobs is NAD+-dependent signaling. In this role, enzymes literally break apart the NAD+ molecule, so, the NAD+ is said to be consumed.
All NAD-dependent signaling pathways cleave NAD+ leaving NAM as a byproduct. The NAM can be recycled to NAD+ in several enzymatic steps, so is said to be “salvageable,” resulting in the salvage pathway name. This is shown in figure 1 where NAM splits off on the pathway to the major NAD+ consuming uses like PARPs, Sirtuins, and CD38.

Figure 2. Nicotinamide (NAM; Niacinamide) Diagram
No matter how NAD+ is made the first time, after NAD+ is consumed in NAD+-dependent signaling reactions, NAM will be generated. Because of this, if a precursor is capable of increasing NAD+, it can also increase NAM. Put another way, NAM levels increase in our bodies no matter which niacin equivalent we start from in a supplement. What’s often overlooked with the newer niacin equivalents (NR, NMN) is that these newer niacins increase NAM in tissues and require it to be salvaged, as does everything with niacin equivalent activity.(1–3)
No matter how NAD+ gets made the first time, it’s going to have to be remade from NAM. When our cells remake NAD+ they don’t care how it got made the first time.
Cells need to constantly replenish the NAD+ pool. Some of this is through building the NAD+ molecule a first time with niacin equivalents in the diet (or supplements). Much of it’s from rebuilding it after it’s been consumed. Let’s get a sense of how much each might contribute.
The daily value (DV) for vitamin B3 is 14 mg/d for women and 16 mg/d for men. But the NAD+ pool is used and replenished several times a day,(4–10) with an estimated 6 to 9 g of NAD+ required daily to match turnover.(11)
This mismatch between a low DV compared to the high daily turnover of NAD+ highlights the importance of salvaging NAM as a means to maintain healthy NAD+ levels.(2, 10, 11) When the salvage pathway is working efficiently, relatively small amounts of dietary vitamin B3 equivalents allow tissues to meet the much larger daily NAD+ needs.
While we could in theory take very large doses of a nutrient that helps build NAD+ as a way to bypass some of the need for salvage, this can come with downsides. Taking very large doses of niacin equivalents (i.e., nutrients that can help us make NAD+), no matter which one—NAM, NA, NR, NMN, Trp—increases risk of unwanted side effects. And high doses of any of these ingredients aren’t free; they tax other metabolic systems. As an example, increasing the dose of niacin equivalents causes more to be methylated (putting extra demands on methylation capacities) and then eliminated in the urine. We think a better approach is to supply a modest amount of a variety of niacin equivalents, while promoting better salvage pathway performance and supporting methylation.
It’s become popular to take high doses of a single niacin equivalent (e.g., NR, NMN) to boost NAD+. We think a better approach is to supply a modest amount of a variety of nutrients that can build NAD+, while promoting better salvage pathway performance and supporting methylation.
The Salvage Pathway Circular Loop
Unlike the de novo pathway (L-tryptophan → NAD+) and the Preiss-Handler pathway (niacin → NAD+), which have a starting point and an end, the salvage pathway is a circle. The progression goes from NAM → NMN → NAD+, which is consumed during NAD+-dependent signaling, leaving NAM as a salvageable byproduct, restarting the salvage loop.
The first enzymatic reaction, starting from NAM, uses nicotinamide phosphoribosyltransferase (Nampt; also called visfatin or pre-B-cell colony-enhancing factor). This is considered the rate-limiting step in the salvage pathway.(12) 5-Phosphoribosyl-1-pyrophosphate (PRPP) is a co-substrate with NAM: The products of the reaction are NMN and pyrophosphate (PPi).(1–3, 13)
Nampt is widely expressed in tissues throughout the body, including adipose tissue, skeletal muscle, brain, kidney, and liver.(14–17) Nampt has high affinity for NAM,(18) which means that it has a natural tendency to want to use NAM if it’s available. Mammals have two different forms of Nampt, intra- and extracellular (iNampt and eNampt, respectively). Both forms catalyze NMN production from NAM.(14) In general, tissues that contain Nampt also contain all salvage pathway enzymes needed to make NAD+ from NAM.
The second step in the salvage pathway converts NMN to NAD+ by a group of ATP-dependent isoenzymes collectively called nicotinate mononucleotide adenylyltransferase (NMNAT). These NMNAT enzymes are shared with the Preiss–Handler pathway. Three forms of this enzyme (NMNAT1, -2, and -3) exist in humans and have distinct tissue and subcellular localizations. Human tissues including brain, heart, kidney, liver, lung, and skeletal muscle express one or more of the NMNAT.
ATP is critical for making NAD+ in the salvage pathway. As is the case with many enzymes, ATP increases the activity of Nampt. It's also required for the NMNAT enzymes, which are responsible for adenylation, also known as AMPylation. This means they attach adenosine monophosphate (AMP) to NMN to form NAD+. ATP is the source of the AMP, so is catabolized in this reaction.(13, 19–22) It’s been suggested that there’s a preferential use of ATP for the biosynthesis of NAD+ when NAM is administered.(23)
ATP is needed to make NAD+ in the salvage pathway. It’s also used in multiple enzymes in the NAD+ metabolome.
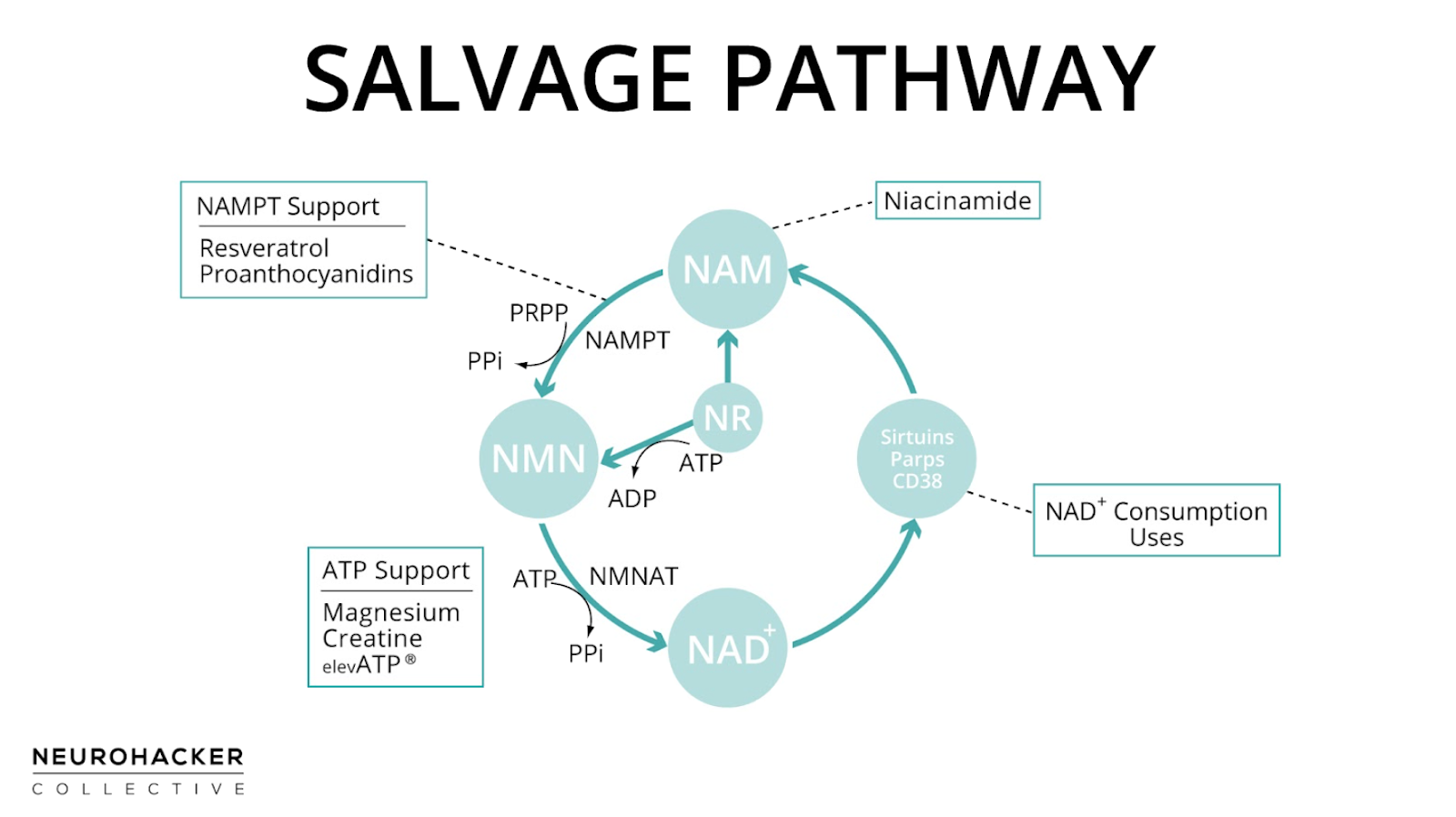
Figure 3. NAM Salvage Pathway with NAD+-Consumption Reactions
Niacinamide - A New Old Way To Make NAD+
Niacinamide (NAM; nicotinamide), along with nicotinic acid (NA; niacin), is one of the two niacins originally classified as vitamin B3. It was discovered in the mid-1930’s and is the 3rd of the B-complex family of vitamins identified, hence its designation as vitamin B3. Unlike NA, NAM does not have lipid-lowering effects and typically does not cause flushing,(24) so is often thought of as the “non-flushing” vitamin B3.
NAM is a cofactor for the structure of all vitamin B3 coenzymes (e.g., NAD+, NADH, NADP, NADPH). NAD+ and NADP are the main forms of niacins in animal products. NAM is cleaved from these larger molecules during digestion and is the dominant absorbed form of niacin from food sources.(25–29) NAM is absorbed from the alimentary canal and enters the bloodstream for distribution to tissues.(5) No matter what’s used to produce NAD+ in the liver, NAM is what’s primarily released from the liver and found in circulation; it’s thought to be the preferred precursor for making NAD molecules in peripheral tissues.(2, 30–32)
NAM markedly increases NAD+ levels in tissues, including brain (NAM crosses the blood-brain barrier),(33–44) eyes,(45) blood,(46–51) kidney,(52) liver(47, 48, 53–63) skeletal muscle,(30), bone marrow,(64), lungs,(65), mammary tissue,(55, 66) pancreas,(67), thyroid,(68) and testes.(69)
It seems to have been forgotten that niacinamide, one of the original vitamin B3’s, (1) increases NAD+, and (2) is the primary building block for NAD+ in most peripheral tissues.
Experimental results suggest that, at physiological concentrations, NAM is used better than NA to produce NAD+ in red blood cells.(12) The same is true in skeletal muscle, while the liver and kidney use both well.(30) In classic experiments, following oral dosing, NAM was a better precursor for NAD+ biosynthesis than NA in all tissues except liver, and to some extent the intestines.(27)
NAM and NA are complementary, since they build NAD+ through different enzyme pathways.(70, 71) NAM also appears to be complementary with de novo synthesis from L-tryptophan (Trp).(72) When administered together, high doses of NAM did not prevent NA from increasing liver NAD+ levels.(5)
It makes sense to supplement both NA and NAM, because responses to each appear to change depending upon the circumstances. NAM, but not NA, elevated liver poly(ADP-ribose) (PARP) (an NAD+-dependent signaling enzyme) in control conditions, but when rats were exposed to a liver toxin, NA, but not NAM supplementation caused a greater accumulation of PARP.(47) In a non-stressed state, NAM was inferior to NA as an NAD+ precursor in the liver,(27) but in animals fed a high-fat diet NAM was a better NAD+ precursor and SIRT1 activator than NA.(61)
These studies suggest circumstances might significantly influence response to niacin equivalents, and that putting all the eggs in one type of vitamin B3 basket, so to speak, might not be the most prudent approach, especially since scientific knowledge about the circumstances and factors that might cause self-regulatory shifts in which NAD+ precursor is preferred in which tissue is extremely limited.
It’s been suggested that supplementing both NA and NAM together may be better than the administration of NA or NAM alone because of their different effects.(71) We agree with this opinion and expect NAM to have additive effects with other NAD+-generating substrates (both Trp and NA). In other words, rather than supporting NAD+ through just one pathway (or one form of niacin equivalent), better benefits might occur (at lower individual doses of each nutrient) when more options are given, because of the complex self-regulatory mechanisms and biological redundancy involved in NAD+ maintenance.
Rather than stress one NAD+ pathway with high amounts of a single NAD+ precursor and hope for the best, we believe it’s better to support all of them, but in moderation. This takes advantage of the functional redundancy for NAD+ generation in many tissues.
There’s 3 ways to make NAD+. This redundancy helps complex systems like our body perform better, especially when faced with real world stresses.
But Doesn’t Niacinamide Block Sirtuins?
One of the misunderstandings we see and hear frequently is the idea that NAM inhibits sirtuins. Sirtuins (SIRTs) are NAD+-consuming deacetylase enzymes, producing NAM as a byproduct. Some enzymes can be slowed down if the product they make builds up. This is called product inhibition. In vitro, NAM (a product of the sirtuin reaction) is a non-competitive sirtuin inhibitor at high concentrations,(72, 73) so shows some degree of product inhibition. But, because this inhibition can occur in cell cultures at high concentrations of NAM, does not mean it would occur in the body at cellular concentrations of NAM. And this inhibition doesn’t always occur in vitro. As an example, several studies using liver cells have found that niacinamide increased sirtuin activity.(42, 74)
In studies where moderate doses of NAM have been given to living animals, in contrast to the predicted in vitro results, NAM increases SIRT activity(61, 75, 76). The reason for the different in vitro and in vivo result is likely because, in our cells and tissues, NAM does not build up to the concentrations needed to inhibit SIRT activity. Instead, it’s salvaged, being fairly rapidly converted to NMN, which then produces NAD+. The result, assuming an efficient flow in the salvage pathway is maintained, is a net positive for SIRT activity. For a more in depth review of this topic we recommend reading “Nicotinamide is an inhibitor of SIRT1 in vitro, but can be a stimulator in cells.”(76)
While niacinamide (NAM) has inhibited sirtuins at high concentrations, it can increase sirtuins when given in moderate amounts.
Support Nampt To Remake NAD+
In addition to supplying NAM as a building block, it’s important that the salvage pathway enzymes be supported. In general, tissue-specific increases in NAD+ levels following either NAM supplementation will be dependent on Nampt expression and activity, since it is the rate-limiting step in the salvage pathway.
Supporting Nampt is also critical if other precursors (NA, NR, NMN, Trp) are used to initially build the NAD+ molecule. This is because NAM is a byproduct of NAD+ consumption uses no matter how the NAD+molecule was originally built. The physiologically produced NAM needs to be salvaged efficiently to remake NAD+.
Because the NAD+ pool is used and replenished several times a day,(4–10) Nampt is believed to be the salvage enzyme most responsible for controlling overall cellular NAD+ amount.(77–80) In general, decreased Nampt expression and/or activity are associated with aging and poorer health, while increased levels appear to be health-protective.
The activity of Nampt enzymes decrease with age, so it’s especially important to follow strategies that support Nampt as we get older.
In studies, Nampt levels generally decline with age in many (but not all) tissues. This results in a decreased ability to produce NAD+ in the affected tissue.(77, 78, 81–84) While this general rule seems to apply, both tissue-specific and region-specific differences can occur. As an example, while brain levels generally decrease with age, in one study Nampt level decreased in cortex and hippocampus, but remained unchanged in cerebellum and striatum brain regions, and increased in microglia. Despite these region-specific changes, the overall effect was a significant decrease in NAD+ in the aged brain.(81)
Chronic inflammation, a common feature in aging, is associated with decreased Nampt.(85, 86) There are also connections between some common age-related health conditions and decreased Nampt activity.(77, 84, 87–91)
Knockout or inhibition of Nampt substantially decreases the ability for a tissue to make NAD+, and produces degenerative and metabolic issues.(17, 42, 92)
In contrast to the associations with decreased Nampt activity with aging and poorer health, increased activity supports better health results. The beneficial adaptive responses to calorie restriction have been linked to Nampt activation (and the subsequent rise in NAD+, sirtuins, and mitochondrial efficiency).(93–95) Exercise training increases Nampt, at least in skeletal muscle.(94, 96, 97) Nampt overexpression in aged mice produced comparable NAD+ levels and the muscle phenotype of young mice.(17) Data also suggest a neuroprotective role for Nampt.(98)
Simply improving the performance of Nampt, without doing anything else to boost NAD+, has made animals biologically younger.
Nampt has a robust circadian rhythm. Circadian transcription factors BMAL and CLOCK control the production of the Nampt enzyme. This is a mechanism for body clock function to direct the circadian synthesis of NAD+ and the activity of NAD+-consuming uses, including SIRTs and PARPs.(99–101) In humans, Nampt levels display a pronounced diurnal rhythm, peaking in the early afternoon.(102) This daytime dominance suggests that supporting increased Nampt activity during the morning and early afternoon might match circadian driven activity.
Resveratrol has increased Nampt in vitro(103–107) and in an animal experiment.(108) In animals, maternal resveratrol intake during lactation positively affects liver protein levels of Nampt in offspring.(109) In the only humans study using resveratrol as an intervention and measuring Nampt, there were non-significant trends towards increased Nampt in skeletal muscle and decreased Nampt in adipose tissue.(110) Grape seed proanthocyanidins are also Nampt-supportive,(111–113) so using grape extracts that contain both resveratrol and proanthocyanidins might provide more support than either in isolation.
In general, strategies that activate AMPK in a tissue would be expected to support better Nampt activity. AMP-Activated Protein Kinase (AMPK) is a cellular energy sensor and functions as an integrating factor for several aging-associated processes. Studies suggest that AMPK activates Nampt, increasing NAD+ recycling, and enhancing SIRT1 activity.(114, 115) We’ll be publishing a future article on AMPK that goes into detail on nutrients and plant extracts that support better AMPK function.
Supporting the NMN to NAD+ Conversion
The NMN to NAD+ uses the NMNAT enzymes: These are the same enzymes used in the Preiss–Handler pathway that creates NAD+ from niacin. So, similar nutritional strategies apply to both.
ATP is needed for the NMNAT enzyme reaction that converts NMN to NAD+. There are a number of nutrients that support ATP production, including cofactors and supportive nutrients for the electron transport chain and the citric acid cycle. Mitochondrial nutrients include CoQ10, PQQ, lipoic acid, and L-carnitine, as examples. We’ll go into more detail of full support for ATP in a subsequent article; however, we want to highlight several ingredients that play substantials roles in ATP function.
ATP is the key molecule needed to support the NMNAT enzymes used to make NAD+ from NMN.
ElevATP® has increased whole blood and intramuscular ATP concentrations in humans,(116, 117) which would be supportive of NMNAT.
Creatine acts as an intracellular buffer for ATP, so is an important part of ATP homeostatic support. Creatine is used in the phosphocreatine (phosphagen) system to regenerate ATP from ADP in tissues. This system is especially important in circumstances where there’s high energy demand. In skeletal muscle, as an example, creatine phosphate (phosphocreatine) acts as a reservoir of high-energy phosphoryl groups that can be readily transferred to ADP to regenerate ATP through the action of the enzyme creatine kinase during higher intensity exercise.(118, 119)
ATP must be bound to a magnesium ion (Mg2+) in order to be biologically active: What is called ATP in cells usually occurs as a magnesium-ATP complex.(120, 121) Because of this, magnesium is essential for ATP performance.
Methylation and Elimination of Niacinamide
Whether NAM is increased directly by giving it as a supplement, or indirectly by giving another precursor that can produce more NAD+, NAM increases in tissues. In fact, in a comparative study, NR increased liver NAM levels to a greater degree than NAM itself did, with niacin (nicotinic acid or NA) having the least effect (see figure 5D in S. A. J. Trammell et al. 2009). One of the other things that increased, shown in figures 5E and 5F in the study, were methylated NAM metabolites.(62)
Excess niacinamide is methylated in the liver to N1-methylnicotinamide (MeNAM), which is excreted in the urine along with its 2- (Me2PY) and 4-pyridone (Me4Py) oxidation products. The proportions of each metabolite produced can vary depending on which niacin equivalent is used and a person’s vitamin B3 status, but the general rule of thumb is that methylated metabolites increase dose-dependently (i.e., higher oral intakes of nutrients that can make NAD+ produce greater increases in methylated metabolites than more modest or lower doses). Because of this relationship, urinary excretion of MeNAM was used to establish the daily value for vitamin B3 intake for adults.(122, 123)
Methylation support is especially important when higher doses of NAM, NR, and NMN are given, but is also important if NA or Trp are used, because anything that builds more NAD+ can increase demands on methylation. Free NAM can be excreted unmetabolized if methylation capacities are exceeded. Moderate doses that are several-fold higher than the daily value for vitamin B3 are below the threshold where free NAM appears in the urine based on research in this area, but still increase methylation demands much more than a daily value amount (i.e., a low dose).(124–128)
Rather than giving high doses of a single NAD+ salvage precursor (which will substantially increase methylated NAM metabolites), we think a better long-term strategy is to give more modest doses of NAD+ precursors, while also supplying extra methylation support. Methylation can be supported with folates (5-MTFR, folinic acid) and vitamin B12 (cobalamins such as methylcobalamin or adenosylcobalamin).
References:
1. G. Magni et al., Cell. Mol. Life Sci. 61, 19–34 (2004).
2. K. L. Bogan, C. Brenner, Annu. Rev. Nutr. 28, 115–130 (2008).
3. C. Dölle, R. H. Skoge, M. R. Vanlinden, M. Ziegler, Curr. Top. Med. Chem. 13, 2907–2917 (2013).
4. Y. Nishizuka, O. Hayaishi, J. Biol. Chem. 238, 3369–3377 (1963).
5. H. Ijichi, A. Ichiyama, O. Hayaishi, J. Biol. Chem. 241, 3701–3707 (1966).
6. G. Elliott, M. Rechsteiner, J. Cell. Physiol. 86, 641–651 (1975).
7. M. Rechsteiner, D. Hillyard, B. M. Olivera, Nature. 259, 695 (1976).
8. M. Rechsteiner, D. Hillyard, B. M. Olivera, J. Cell. Physiol. 88, 207–217 (1976).
9. G. T. Williams, K. M. Lau, J. M. Coote, A. P. Johnstone, Exp. Cell Res. 160, 419–426 (1985).
10. Y. Yang, A. A. Sauve, Biochim. Biophys. Acta. 1864, 1787–1800 (2016).
11. A. Chiarugi, C. Dölle, R. Felici, M. Ziegler, Nat. Rev. Cancer. 12, 741–752 (2012).
12. V. Micheli, H. A. Simmonds, S. Sestini, C. Ricci, Arch. Biochem. Biophys. 283, 40–45 (1990).
13. A. Nikiforov, V. Kulikova, M. Ziegler, Crit. Rev. Biochem. Mol. Biol. 50, 284–297 (2015).
14. J. R. Revollo et al., Cell Metab. 6, 363–375 (2007).
15. L. R. Stein et al., J. Neurosci. 34, 5800–5815 (2014).
16. J. Camacho-Pereira et al., Cell Metab. 23, 1127–1139 (2016).
17. D. W. Frederick et al., Cell Metab. 24, 269–282 (2016).
18. E. S. Burgos, V. L. Schramm, Biochemistry. 47, 11086–11096 (2008).
19. M. Schweiger et al., FEBS Lett. 492, 95–100 (2001).
20. F. Berger, C. Lau, M. Ziegler, Proc. Natl. Acad. Sci. U. S. A. 104, 3765–3770 (2007).
21. C. Lau, M. Niere, M. Ziegler, Front. Biosci. . 14, 410–431 (2009).
22. A. A.-B. Badawy, Int. J. Tryptophan Res. 10, 1178646917691938 (2017).
23. C. Bernofsky, Mol. Cell. Biochem. 33, 135–143 (1980).
24. M. Knip et al., Diabetologia. 43, 1337–1345 (2000).
25. J. B. Turner, D. E. Hughes, Exp. Physiol. 47, 107–123 (1962).
26. C. Streffer, J. Benes, Eur. J. Biochem. 21, 357–362 (1971).
27. P. B. Collins, S. Chaykin, J. Biol. Chem. 247, 778–783 (1972).
28. L. M. Henderson, C. J. Gross, J. Nutr. 109, 654–662 (1979).
29. L. M. Henderson, C. J. Gross, J. Nutr. 109, 646–653 (1979).
30. K. Shibata, T. Hayakawa, K. Iwai, Agric. Biol. Chem. 50, 3037–3041 (1986).
31. M. F. Murray, Clin. Infect. Dis. 36, 453–460 (2003).
32. S.-I. Imai, FEBS Lett. 585, 1657–1662 (2011).
33. R. Spector, Neurochem. Res. 12, 27–31 (1987).
34. L. K. Klaidman, S. K. Mukherjee, T. P. Hutchin, J. D. Adams, Neurosci. Lett. 206, 5–8 (1996).
35. F. J. Wan, H. C. Lin, B. H. Kang, C. J. Tseng, C. S. Tung, Brain Res. Bull. 50, 167–171 (1999).
36. J. Yang et al., Neurosci. Lett. 333, 91–94 (2002).
37. F. Sadanaga-Akiyoshi et al., Neurochem. Res. 28, 1227–1234 (2003).
38. L. Klaidman et al., Pharmacology. 69, 150–157 (2003).
39. R. Spector, C. E. Johanson, J. Neurochem. 103, 425–438 (2007).
40. D. Liu, R. Gharavi, M. Pitta, M. Gleichmann, M. P. Mattson, Neuromolecular Med. 11, 28–42 (2009).
41. C. S. Siegel, L. D. McCullough, Neuroscience. 237, 223–231 (2013).
42. J. Li et al., Biochim. Biophys. Acta. 1853, 2929–2936 (2015).
43. F. Li et al., Proc. Natl. Acad. Sci. U. S. A. 113, 13450–13455 (2016).
44. C. Wang et al., Neural Plast. 2017, 7019803 (2017).
45. P. A. Williams et al., Science. 355, 756–760 (2017).
46. B. Petrack, P. Greengard, H. Kalinsky, J. Biol. Chem. 241, 2367–2372 (1966).
47. T. M. Jackson, J. M. Rawling, B. D. Roebuck, J. B. Kirkland, J. Nutr. 125, 1455–1461 (1995).
48. M. M. ApSimon, J. M. Rawling, J. B. Kirkland, J. Nutr. 125, 1826–1832 (1995).
49. K. Majamaa, H. Rusanen, A. M. Remes, J. Pyhtinen, I. E. Hassinen, Life Sci. 58, 691–699 (1996).
50. K. Majamaa, H. Rusanen, A. Remes, I. E. Hassinen, Mol. Cell. Biochem. 174, 291–296 (1997).
51. Y. Takahashi et al., Kidney Int. 65, 1099–1104 (2004).
52. M. T. Tran et al., Nature. 531, 528–532 (2016).
53. R. Van Reen, M. Quesada, Biochem. Biophys. Res. Commun. 24, 56–60 (1966).
54. O. Hayaishi, H. Ijichi, A. Ichiyama, Adv. Enzyme Regul. 5, 9–22 (1967).
55. S. Pinder, A. L. Greenbaum, Biochem. J. 102, 20C–21C (1967).
56. J. B. Clark, S. Pinder, Biochem. J. 114, 321–330 (1969).
57. J. T. MacGregor, A. Burkhalter, Biochem. Pharmacol. 22, 2645–2658 (1973).
58. C. L. Baum, J. Selhub, I. H. Rosenberg, Biochem. J. 204, 203–207 (1982).
59. G. M. McCreanor, D. A. Bender, Br. J. Nutr. 56, 577–586 (1986).
60. V. Batra, B. Kislay, Mutat. Res./Fundam. Mol. Mech. Mutag. 749, 28–38 (2013).
61. S. J. Yang et al., J. Nutr. Biochem. 25, 66–72 (2014).
62. S. A. J. Trammell et al., Nat. Commun. 7, 12948 (2016).
63. S. Á. Mejía et al., Eur. J. Pharmacol. 818, 499–507 (2018).
64. A. C. Boyonoski et al., J. Nutr. 132, 115–120 (2002).
65. A. Nagai et al., Exp. Lung Res. 20, 263–281 (1994).
66. A. L. Greenbaum, S. Pinder, Biochem. J. 107, 55–62 (1968).
67. H. Tjälve, E. Wilander, Acta Endocrinol. . 83, 357–364 (1976).
68. C. H. Bastomsky, V. Abbassi, J. M. McKenzie, Endocrinology. 83, 79–85 (1968).
69. L. F. Lin, L. M. Henderson, J. Biol. Chem. 247, 8023–8030 (1972).
70. N. Hara et al., J. Biol. Chem. 282, 24574–24582 (2007).
71. K. Shibata, T. Fukuwatari, C. Suzuki, J. Nutr. Sci. Vitaminol. . 60, 86–93 (2014).
72. T. Fukuwatari, K. Shibata, Int. J. Vitam. Nutr. Res. 77, 255–262 (2007).
73. J. L. Avalos, K. M. Bever, C. Wolberger, Mol. Cell. 17, 855–868 (2005).
74. C. Shen et al., Nutr. Res. 40, 40–47 (2017).
75. M. Pajk et al., Biogerontology. 18, 593–600 (2017).
76. E. S. Hwang, S. B. Song, Cell. Mol. Life Sci. 74, 3347–3362 (2017).
77. J. Yoshino, K. F. Mills, M. J. Yoon, S.-I. Imai, Cell Metab. 14, 528–536 (2011).
78. L. Mouchiroud et al., Cell. 154, 430–441 (2013).
79. A. Garten, S. Petzold, A. Körner, S.-I. Imai, W. Kiess, Trends Endocrinol. Metab. 20, 130–138 (2009).
80. S.-I. Imai, Curr. Pharm. Des. 15, 20–28 (2009).
81. L.-Y. Liu et al., PLoS One. 7, e44933 (2012).
82. A. P. Gomes et al., Cell. 155, 1624–1638 (2013).
83. L. R. Stein, S.-I. Imai, EMBO J. 33, 1321–1340 (2014).
84. D. Ghosh, K. R. Levault, G. J. Brewer, Aging Cell. 13, 631–640 (2014).
85. T. Singh, A. B. Newman, Ageing Res. Rev. 10, 319–329 (2011).
86. S. Imai, J. Yoshino, Diabetes Obes. Metab. 15 Suppl 3, 26–33 (2013).
87. C.-P. Hsu, S. Oka, D. Shao, N. Hariharan, J. Sadoshima, Circ. Res. 105, 481–491 (2009).
88. T. B. Dahl et al., J. Clin. Endocrinol. Metab. 95, 3039–3047 (2010).
89. S. Jukarainen et al., J. Clin. Endocrinol. Metab. 101, 275–283 (2016).
90. X. Wang, H. Li, S. Ding, Sci. Rep. 6, 32416 (2016).
91. X. Wang et al., Cell Rep. 20, 2184–2200 (2017).
92. K. L. Stromsdorfer et al., Cell Rep. 16, 1851–1860 (2016).
93. E. van der Veer et al., J. Biol. Chem. 282, 10841–10845 (2007).
94. C. Cantó et al., Cell Metab. 11, 213–219 (2010).
95. J. Song et al., J. Gerontol. A Biol. Sci. Med. Sci. 69, 44–57 (2014).
96. S. R. Costford et al., Am. J. Physiol. Endocrinol. Metab. 298, E117–26 (2010).
97. E. Koltai et al., Mech. Ageing Dev. 131, 21–28 (2010).
98. Z. Jing et al., J. Cereb. Blood Flow Metab. 34, 1613–1621 (2014).
99. Y. Nakahata, S. Sahar, G. Astarita, M. Kaluzova, P. Sassone-Corsi, Science. 324, 654–657 (2009).
100. K. M. Ramsey et al., Science. 324, 651–654 (2009).
101. H.-C. Chang, L. Guarente, Cell. 153, 1448–1460 (2013).
102. C. Benedict et al., J. Clin. Endocrinol. Metab. 97, E218–22 (2012).
103. S. V. de Kreutzenberg et al., Diabetes. 59, 1006–1015 (2010).
104. S. Schuster et al., PLoS One. 9, e91045 (2014).
105. K. C. Morris-Blanco, C. H. Cohan, J. T. Neumann, T. J. Sick, M. A. Perez-Pinzon, J. Cereb. Blood Flow Metab. 34, 1024–1032 (2014).
106. P. Huang et al., Oncotarget. 6, 10812–10824 (2015).
107. F. Lan, K. A. Weikel, J. M. Cacicedo, Y. Ido, Nutrients. 9 (2017), doi:10.3390/nu9070751.
108. Y. Lee et al., Comp. Biochem. Physiol. C. Toxicol. Pharmacol. 208, 71–76 (2018).
109. M. Tanaka et al., Biochem Biophys Rep. 9, 173–179 (2017).
110. J. Yoshino et al., Cell Metab. 16, 658–664 (2012).
111. A. Ribas-Latre et al., Sci. Rep. 5, 10954 (2015).
112. A. Ribas-Latre et al., Mol. Nutr. Food Res. 59, 865–878 (2015).
113. G. Aragonès et al., Sci. Rep. 6, 24977 (2016).
114. M. Fulco et al., Dev. Cell. 14, 661–673 (2008).
115. C. Cantó et al., Nature. 458, 1056–1060 (2009).
116. T. Reyes-Izquierdo et al., J Aging Res Clin Practice. 2, 178–184 (2013).
117. T. Reyes-Izquierdo, C. Shu, R. Argumedo, B. Nemzer, Z. Pietrzkowski, J Aging Res Clin Pract. 3, 56–60 (2014).
118. P. L. Greenhaff, J. Physiol. 537, 657–657 (2001).
119. L. Guimarães-Ferreira, Einstein . 12, 126–131 (2014).
120. R. M. Touyz, Front. Biosci. 9, 1278–1293 (2004).
121. K. Pasternak, J. Kocot, A. Horecka, Journal of Elementology. 15, 601–616 (2010).
122. J. E. Mrochek, R. L. Jolley, D. S. Young, W. J. Turner, Clin. Chem. 22, 1821–1827 (1976).
123. Institute of Medicine (US) Standing Committee on the Scientific Evaluation of Dietary Reference Intakes and its Panel on Folate, Other B Vitamins, and Choline, Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline (National Academies Press (US), Washington (DC), 2012).
124. SHIBATA, K, Vitam. Horm. 66, 309–314 (1992).
125. M. R. Stratford, M. F. Dennis, J. Chromatogr. 582, 145–151 (1992).
126. K. Aoyama et al., J. Neural Transm. 107, 985–995 (2000).
127. K. Shibata, J. Nutr. 119, 892–895 (1989).
128. S. Katsumi, S. Hisako, T. Hiroshi, Biosci. Biotechnol. Biochem. 60, 1204–1206 (1996).











Dr. Ben Lynch