
Key learning objectives
Find out how metabolism and immunity influence each other
Understand the role of mitochondria in immunometabolism
Discover how mitochondria participate in immune cell responses
Understand the importance of mitochondria for healthy immunity
What Is Immunometabolism?
Metabolism is the set of chemical reactions that sustain the activity of cells and organs. All biological processes are supported by metabolic reactions. Immunity is no exception: the activation, growth, proliferation of immune cells, the enactment of immune functions, and the return to homeostasis are all dependent on metabolism and on its fast adaptation in the face of new immune challenges.
Immune responses are energetically demanding processes as they require large amounts of cellular energy and metabolic building blocks to sustain the fast proliferation and the activity of all immune cells, as well as the production of all chemical mediators involved. The management and allocation of metabolic resources is therefore a key element of immune responses. This association between metabolic regulation and adaptation and immune responses is called immunometabolism.
Mitochondria, the cellular organelles at the core of all metabolic processes, are key elements in immunometabolism. Not only are they generators of cell energy, but also important signaling hubs in all cell types. Mitochondria thus hold central stage in the crosstalk between metabolic and immune signaling pathways.
Immunometabolism is the crosstalk between metabolic and immunological processes. It is an emerging field of scientific research.
Mitochondria Are Powerhouses of Immunity
The energy molecule of cells is called adenosine triphosphate, better known as ATP. ATP is produced in mitochondria using energy extracted from food in metabolic pathways that converge in mitochondria: the breakdown of glucose via glycolysis and of fatty acids via fatty acid oxidation (also known as beta oxidation). Both yield acetyl-CoA, which enters the citric acid cycle in mitochondria to be further metabolized. Together, these three processes extract electrons from (i.e., oxidize) glucose and fatty acids, which are then carried by NAD+ (as NADH) and FAD+ (as FADH2) to the mitochondrial electron transport chain to generate ATP through oxidative phosphorylation [1,2]. (We invite you to read the linked articles if you want to learn more about these processes.)
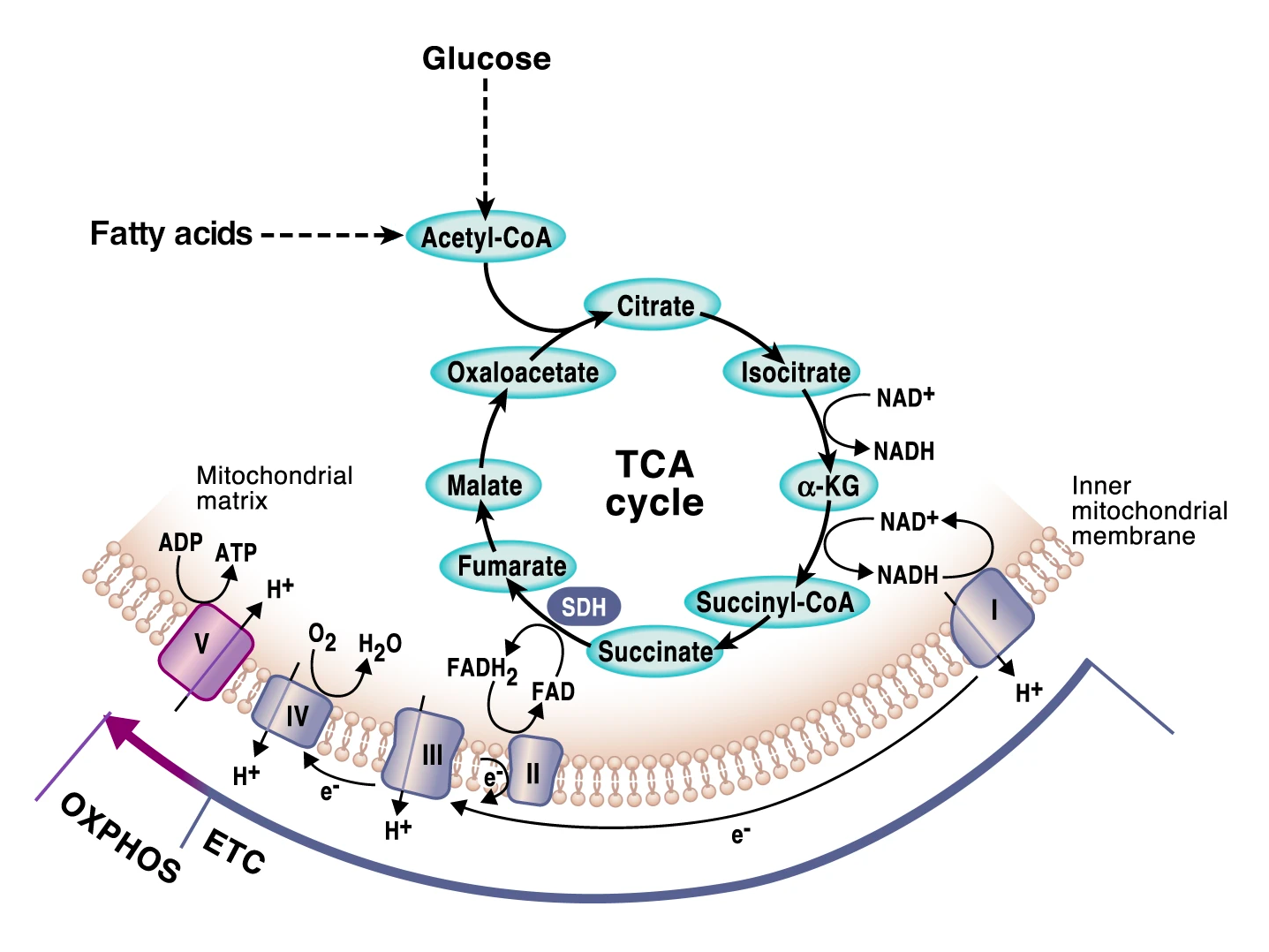
Figure 1:Cell energy pathways in mitochondria.
Source: Adapted from Martínez-Reyes & Chandel, Nat. Commun. 11:102 (2020).
License CC BY 4.0.
ATP is the molecule that provides the energy required to support cell functions. In the absence of immune challenges, immune cells are mostly in a state of metabolic rest known as quiescence. Long-term survival in the quiescence state is supported by ATP produced by the steady metabolic breakdown of large molecules (i.e., macromolecules).
Upon activation, immune cells shift to a metabolic state in which ATP is being used at a high rate to power the assembly of all the macromolecules that are required for immune function. When an immune response is being mounted, it’s ATP that powers the construction of new cellular structures, and ultimately, new immune cells; it’s also ATP that powers the synthesis of chemical mediators by immune cells. In other words, ATP provides the energy required for the proliferation and activity of immune cells. Because mitochondria are the main site of ATP generation, they are the main providers of bioenergetic support for immune cell function [3].
Mounting an immune response typically entails extensive changes in gene expression which support the fast division and growth of new cells and the execution of new functions, such as producing and releasing signaling molecules and migration to sites of immune challenges. When an immune response is triggered, the transition from quiescence into activity creates a shift in the metabolic requirements that mitochondria will need to fulfill by adjusting the allocation of metabolic resources to different pathways according to cellular needs [3].
The fast proliferation of functional immune cells will entail constructing every cell structure in every new cell from scratch. It’s a huge metabolic endeavor that requires mitochondria working at full capacity.
During immune cell activation, mitochondria support metabolic shifts from a state of metabolic rest to a state of high energy demand.
Immune cells may have to, relatively speaking, travel great distances from where they would be resting in a quiescent state to where they are needed to deal with challenges. ATP plays a critical role in the migration of immune cells in several ways. White blood cells need to increase ATP production to power their migration [4]. They do this by rearrangement of mitochondrial networks within immune cells so that the high amounts of ATP required for movement are produced in the areas where it is needed to help the immune cells move quickly [4,5]. ATP is also involved in recruiting immune cells. Stressed and damaged cells release ATP. This extracellular ATP can be detected by immune cells, acting as a signal that alerts them to move rapidly to locations in the body where immune work is required [6,7].
Mitochondria are the main generators of ATP, as we’ve seen, but they are also essential in providing building blocks for the synthesis of the molecules required for cellular proliferation. For example, the citric acid cycle is a source of precursors for the production of DNA and proteins, both of which are indispensable for cell division. Mitochondria are responsible for managing metabolic pathways to meet cellular needs, either increasing the production of ATP or increasing the availability of metabolic precursors for macromolecule synthesis via the citric acid cycle [1,2]. By doing so, mitochondria also help to control the various metabolic decision points that determine immune cell function [8].
Mitochondria Are Immunometabolic Signaling Platforms
Mitochondria are also important cellular signaling hubs that regulate signaling pathways and the gene expression profiles that drive the biological functions of immune cells [9]. Through their molecules, machinery, and processes—mitochondrial DNA (mtDNA), mitochondrial reactive oxygen species (mtROS), metabolic pathways, signaling pathways, etc.—mitochondria can regulate all steps of immune cell activity, from activation to differentiation, proliferation, migration, and survival [10].

Figure 2: Essential signaling functions of mitochondria.
Source: Martínez-Reyes & Chandel, Nat. Commun. 11:102 (2020). License CC BY 4.0.
Mitochondrial ROS (mtROS) are among the most important signaling molecules produced by mitochondria. The mitochondrial electron transport chain (ETC), the site of ATP production, is also the major site of mtROS production. MtROS have key signaling functions in many important cellular processes, including those that underlie immune responses. They diffuse from mitochondria to the cytosol and reach the cell nucleus, where they modulate the activity of transcription factors. These are proteins that bind to specific DNA sequences to control the rate of transcription of genetic information, resulting in changes in gene expression that lead to changes in the activity and function of cells. Most immune cells are reliant on mtROS signaling for the generation of an appropriate immune response [9,11].
Mitochondrial metabolites can also modulate gene expression. For example, the citric acid cycle intermediates α-ketoglutarate and succinate are able to influence gene expression through epigenetic modifications (i.e., changes in gene activity through the addition of chemical groups without altering the DNA sequence, such as methylations or acetylations, for example). The same is true for acetyl-CoA, which, as mentioned above, is the molecule produced in the oxidation of glucose and fatty acids and metabolized in the citric acid cycle. Acetyl-CoA is used for the acetylation of histones, which are proteins that provide structural support to chromosomes and that can influence the rate of gene transcription. Nuclear acetyl-CoA levels are determined by its mitochondrial availability, which means that changes in the flux of mitochondrial metabolic pathways can alter the availability of acetyl-CoA and thereby inhibit or promote epigenetic changes that regulate immune cell function [8].

Figure 3: Epigenetic regulation of gene expression by citric acid cycle intermediates.
Source: Martínez-Reyes & Chandel, Nat. Commun. 11:102 (2020). License CC BY 4.0.
Mitochondrial DNA can also act as a signaling molecule. For example, when mitochondria are damaged, mtDNA is released into the cytosol of cells, where it acts as a signal from the mitochondria to the nucleus alerting the cell that significant damage is occurring. MtDNA is capable of activating major innate immune signaling pathways that lead to the activation of immune system communication genes [9,12].
Mitochondrial molecules and pathways are able to modulate all steps of immune responses, from immune cell activation and proliferation to the enactment of immune responses.
How Mitochondria Support Macrophage Responses
Macrophages are a great example of how mitochondrial metabolic pathways can adapt to, and support, different types of challenges and responses. Macrophages are cells of the innate immune system that “eat” large molecules and even cells—that’s where their name comes from, the Greek words for large (makros) and eating (phagein). Macrophages eliminate foreign or damaged cells through phagocytosis, i.e., their ingestion and digestion in specialized intracellular structures through the actions of highly reactive molecules—ROS, reactive nitrogen species (RNS), and enzymes.
There are two categories of macrophages—M1 macrophages are involved in initiating the early stages of an immune response and M2 macrophages are involved in the tissue remodeling and repair that takes place in the later stages of an immune response. Macrophages become either M1 or M2 in reaction to stimuli and signals in their environment; this process is known as macrophage polarization [13].
These two types of macrophages have a notorious difference in their citric acid cycle that is directly linked to their functions. In M1 macrophages, the citric acid cycle is “broken” at two points: it has two enzymatic blocks at the reactions catalyzed by isocitrate dehydrogenase (IDH) and succinate dehydrogenase (SDH) [14].
The block at IDH, which converts isocitrate to α-ketoglutarate, causes a build-up of the isocitrate precursor cis-aconitate. This molecule is used in M1 macrophages to produce a molecule with antibacterial properties called itaconate [14,15]. The inhibition of IDH also causes an accumulation of citrate, which is transported from the mitochondria to the cytosol where it supports responses in M1 macrophages via ROS and nitric oxide (NO) signaling [9,14,16].
The block at SDH is associated with changes in the ETC. SDH, which converts succinate to fumarate, is also part of complex II of the ETC. SDH typically receives electrons from succinate and passes them on to complex III for forward ETC flux. Because of the SDH block, reverse electron transfer (RET) in the ETC is promoted, with electrons being transferred from complex II to complex I, which in turn generates mtROS. MtROS then activate the transcription factor hypoxia-inducible factor 1α (HIF1α) and HIF-1α-dependent gene expression, including the production of immune communication molecules.
These metabolic alterations thus repurpose mitochondria from ATP synthesis to mtROS production to promote a state of immune responsiveness, with SDH as a key control point in host defense [9,17]. Transiently producing this state is an essential part of immune system performance. But prolonging it can lead to excessive ROS production and disproportionate immune responses. To prevent these potentially damaging effects, macrophages use the metabolite produced in the IDH block, itaconate. Itaconate inhibits SDH and decreases the generation of mtROS derived from succinate-induced RET at complex I. Consequently, it limits HIF-1α activation and regulates the production of cytokines, keeping the activity of M1 macrophages within beneficial limits [9,18].
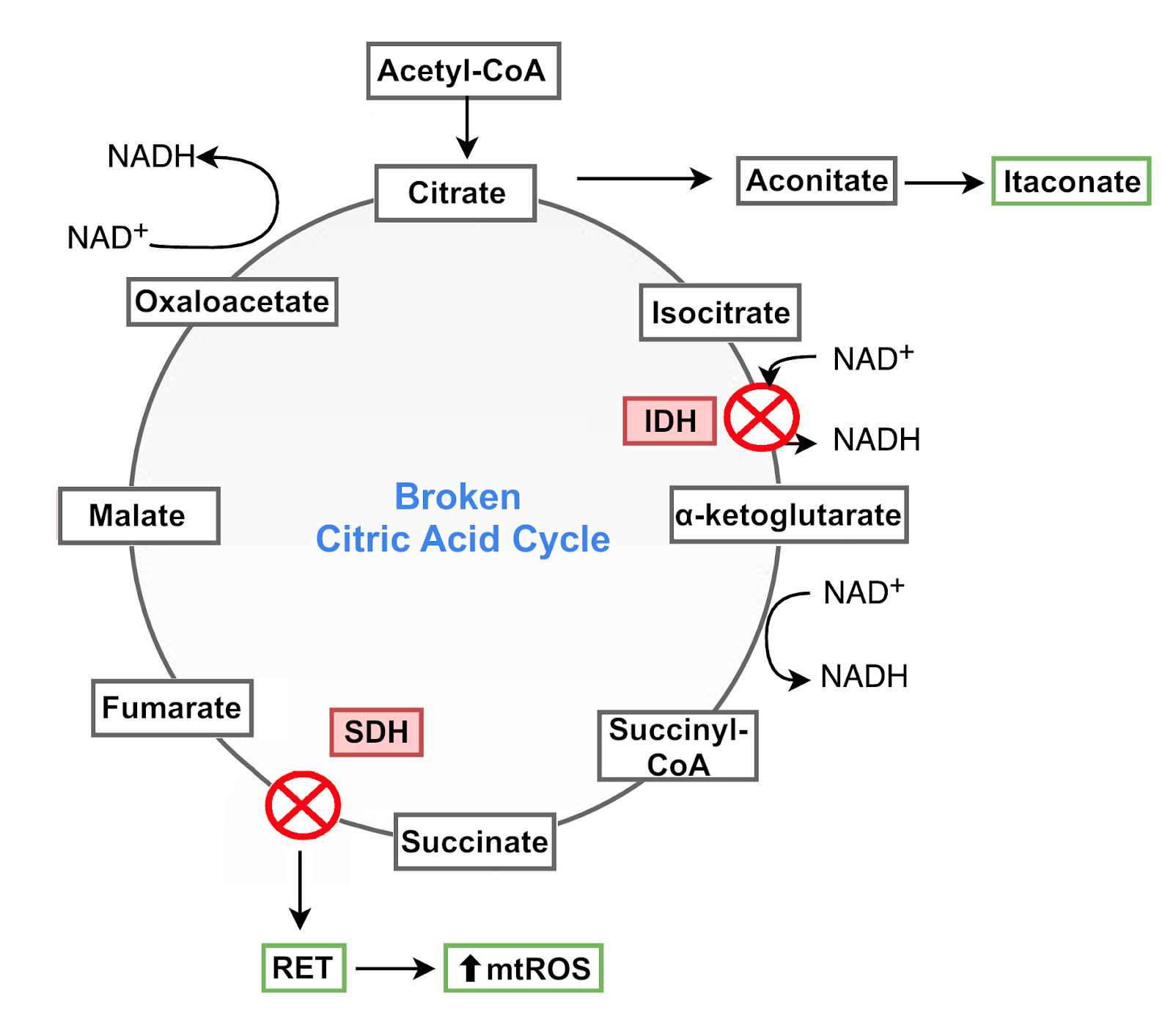
Figure 4: Broken citric acid cycle in M1 macrophages.
Despite these two blocks, the flow of the citric acid cycle is maintained through the reintroduction into the cycle of α‑ketoglutarate and fumarate derived from other metabolic pathways [14].
Unlike M1 macrophages, M2 macrophages have an intact citric acid cycle and a metabolic program heavily fueled by mitochondrial fatty acid oxidation (FAO), which is boosted via signaling by AMPK and PPAR-δ [10,19]. The enhancement of FAO in M2 macrophages shifts the functions of macrophages and thus supports the type of macrophage profile desired during the resolution phase of an immune response [9]. M2 macrophages also display increased mitochondrial biogenesis—essentially the process of producing a bigger mitochondrial network—owing to the expression PGC1β [20], which is known to be a consequence of AMPK signaling [21].
How Mitochondria Support T Cell Responses
T cells, which are cells of the adaptive immune system, provide another good example of how mitochondria support the development of immune responses. The metabolism of T cells varies not only with their activation state (quiescent vs active), but also with their developmental state (naive vs differentiated) and function (e.g., helper T cell, effector T cell). When quiescent naive T cells are activated, they undergo rapid proliferation and differentiation. This is accompanied by a substantial increase in glycolysis and mitochondrial metabolism through the citric acid cycle to support the production of ATP and, importantly, provide precursors needed to meet the extremely high biosynthetic demands of growing and proliferating cells.
T cell activation is accompanied by drastic changes in mitochondria, with increased mitochondrial biogenesis that gives rise to distinct populations of mitochondria with specialized functions and metabolic signatures better suited to support T cell proliferation and survival [22].
Changes in mitochondrial dynamics (adjustments in mitochondrial size and shape that occur through fusion and fission of mitochondria), cristae organization (mitochondrial cristae are infolding of the ETC-containing inner mitochondrial membrane that increase its area), and ETC architecture also allow mitochondria to adjust their activity to the metabolic requirements of each T cell subpopulation.
Fused, elongated mitochondria tend to have a more efficient ETC and OXPHOS, which is important for cell survival in times of stress [23,24]; fission, on the other hand, generates fragmented mitochondria with increased ROS production, which may be important during T cell activation and proliferation, and enhances mitophagy [25,26]. Naive T cells have fragmented, round mitochondria [22]; memory T cells (Tm cells) have elongated mitochondria with increased mitochondrial mass; effector T cells (Teff cells) have increased mitochondrial fission and more punctate mitochondria with looser cristae [27].
How Metabolism and The Immune System Influence Each Other
We’ve seen that mitochondrial metabolism and signaling are at the core of immune responses. But there are other ways through which metabolism and immunity influence each other.
Fatty acids can act as immunomodulatory molecules, with some types of fatty acids (e.g., saturated fatty acids) stimulating the production of immune signaling mediators that are more characteristic of an active immune response[28,29], and other types (e.g., omega-3 fatty acids) influencing communication molecules in ways that are more consistent with a quiescent immune system [30]. This specificity of how different types of fatty acids are used by immune cells implies that the composition of our diet can have a direct influence on immune signaling and performance.
The gut microbiota, which has an important influence on our metabolism and immunity, also plays a part in the immunometabolic crosstalk. For example, short-chain fatty acids, which are byproducts of fermentation by the gut microbiota, can stimulate the differentiation of T cells into Treg cells, which are a subpopulation of T cells specialized in keeping immune responses in check [31–33].
And because the immunometabolic crosstalk is bidirectional, immunity can also influence metabolism. In fact, metabolic problems are known to occur in the setting of active immune responses. For example, molecules produced by macrophages can induce insulin resistance and immune challenges have been associated with decreased insulin signaling. In fact, macrophages and other immune cells, through an abnormal production of signaling molecules, are known to play an important role in obesity and other metabolic dysfunctions [34–39]. In turn, obesity has been associated with changes in immune responses, including an abnormal activity of innate immune cells [40] and a decrease in T cell responsiveness [41].
Why Supporting Mitochondria Is Important For Immune Health
As we’ve seen, the overlap between metabolic and immune signaling pathways can quickly create a vicious spiral in which altered metabolic states have a negative impact on immune function, and altered immunity has a negative impact on metabolism. Maintaining a healthy metabolic function can thus contribute to our immune health, which in turn can contribute to our metabolic health.
Because of the central role of mitochondria in metabolism, they are major determinants of metabolic health and, consequently, immune health. Accordingly, mitochondrial dysfunction (ROS production due to inhibition of ETC complexes, mtDNA release due to mitochondria damage, etc) has been linked to unhealthy immune signaling through the activation of signaling pathways (e.g., the NLRP3 inflammasome) known to have damaging effects in many different tissues and organs of the human body [42–45].
Furthermore, mitochondrial dysfunction is one of the hallmarks of aging [46]. Aging-related mitochondrial dysfunction is associated with increased mtROS production and release of mtDNA, which have been linked to the increased production of cytokines associated with the inflammaging observed in aging. Declines in mitochondrial function have also been linked to aging-related declines in immunity and T-cell responses [47].
Therefore, supporting healthy mitochondrial function may help our immune cells stay fit and intelligent, so they can respond properly to immune challenges. Because the performance of the immune system gradually declines during aging—a process called immunosenescence—supporting healthy mitochondria may be even more important as we grow older.
How The Support of Metabolism and Immunity May Be Synergistic
Throughout this article, we learned of some of the many ways in which energy metabolism and immunity are connected. It’s fairly clear that metabolic health is essential for a healthy immune function and that immune health is essential for a healthy metabolic function.
Qualia Immune was designed to comprehensively support a smarter, fitter immune system. You probably won’t be surprised to find out that mitochondrial support was one of Qualia’s considerations when formulating it. In our blog post describing how it works (see Qualia Immune - The Science Behind the Formula & Immune System Intelligence), there's a subsection titled “Immune Thinking Takes Energy” that discusses this area specifically. Many of the ingredients included in Qualia Immune—Resveratrol [48–52], Broccoli Sprout Powder (Sulforaphane) [53–58], Palmitoylethanolamide [59–62], Spirulina Extract [63–67], Reishi Mushroom [68–71], Panax ginseng [72–76], Cranberry Fruit [77–81], Fucoidan (from Fucus vesiculosus and Undaria pinnatifida) [82–87], Vitamin D3 [88–92], Zinc [93–97], Selenium [98–102], and N-Acetyl-L-cysteine [103–106]—support the immune system and mitochondrial performance.
At Qualia, we believe that helping immune cells respond to the increased energy demands that occur during immune challenges contributes to faster and more intelligent immune responses. We hope that, after reading this blog post, you’ll appreciate why we made sure Qualia Immune offered substantial mitochondrial support.
We also want to mention that Qualia Life may be a good complement to Qualia Immune, especially when even greater mitochondrial support is desired. Main targets of Qualia Life include enhancing mitochondrial function and supporting the main cell energy pathways. We invite you to learn more about the design of Qualia Life in the article Qualia Life: Putting The Healthy Aging Puzzle Together.
References
[1]J.M. Berg, J.L. Tymoczko, G.J. Gatto, L. Stryer, eds., Biochemistry, 8th ed, W.H. Freeman and Company, 2015.
[2]D.L. Nelson, M.M. Cox, Lehninger Principles of Biochemistry, 7th Edition, W. H. Freeman and Company, 2017.
[3]A. Angajala, S. Lim, J.B. Phillips, J.-H. Kim, C. Yates, Z. You, M. Tan, Front. Immunol. 9 (2018) 1605.
[4]F.M. Marelli-Berg, M. Jangani, J. Leukoc. Biol. 104 (2018) 285–293.
[5]S. Campello, R.A. Lacalle, M. Bettella, S. Mañes, L. Scorrano, A. Viola, J. Exp. Med. 203 (2006) 2879–2886.
[6]P.J. Sáez, P. Vargas, K.F. Shoji, P.A. Harcha, A.-M. Lennon-Duménil, J.C. Sáez, Sci. Signal. 10 (2017).
[7]C. Ledderose, K. Liu, Y. Kondo, C.J. Slubowski, T. Dertnig, S. Denicoló, M. Arbab, J. Hubner, K. Konrad, M. Fakhari, J.A. Lederer, S.C. Robson, G.A. Visner, W.G. Junger, J. Clin. Invest. 128 (2018) 3583–3594.
[8]M.M. Mehta, S.E. Weinberg, N.S. Chandel, Nat. Rev. Immunol. 17 (2017) 608–620.
[9]E.L. Mills, B. Kelly, L.A.J. O’Neill, Nat. Immunol. 18 (2017) 488–498.
[10]A. Angajala, S. Lim, J.B. Phillips, J.-H. Kim, C. Yates, Z. You, M. Tan, Front. Immunol. 9 (2018) 1605.
[11]E.L. Pearce, E.J. Pearce, Immunity 38 (2013) 633–643.
[12]A. Mohanty, R. Tiwari-Pandey, N.R. Pandey, J. Cell Commun. Signal. 13 (2019) 303–318.
[13]P.J. Murray, Annu. Rev. Physiol. 79 (2017) 541–566.
[14]A.K. Jha, S.C.-C. Huang, A. Sergushichev, V. Lampropoulou, Y. Ivanova, E. Loginicheva, K. Chmielewski, K.M. Stewart, J. Ashall, B. Everts, E.J. Pearce, E.M. Driggers, M.N. Artyomov, Immunity 42 (2015) 419–430.
[15]A. Michelucci, T. Cordes, J. Ghelfi, A. Pailot, N. Reiling, O. Goldmann, T. Binz, A. Wegner, A. Tallam, A. Rausell, M. Buttini, C.L. Linster, E. Medina, R. Balling, K. Hiller, Proc. Natl. Acad. Sci. U. S. A. 110 (2013) 7820–7825.
[16]V. Infantino, P. Convertini, L. Cucci, M.A. Panaro, M.A. Di Noia, R. Calvello, F. Palmieri, V. Iacobazzi, Biochem. J 438 (2011) 433–436.
[17]E. Mills, L.A.J. O’Neill, Trends Cell Biol. 24 (2014) 313–320.
[18]V. Lampropoulou, A. Sergushichev, M. Bambouskova, S. Nair, E.E. Vincent, E. Loginicheva, L. Cervantes-Barragan, X. Ma, S.C.-C. Huang, T. Griss, C.J. Weinheimer, S. Khader, G.J. Randolph, E.J. Pearce, R.G. Jones, A. Diwan, M.S. Diamond, M.N. Artyomov, Cell Metab. 24 (2016) 158–166.
[19]M. Kemmerer, F. Finkernagel, M.F. Cavalcante, D.S.P. Abdalla, R. Müller, B. Brüne, D. Namgaladze, PLoS One 10 (2015) e0130893.
[20]D. Vats, L. Mukundan, J.I. Odegaard, L. Zhang, K.L. Smith, C.R. Morel, R.A. Wagner, D.R. Greaves, P.J. Murray, A. Chawla, Cell Metab. 4 (2006) 13–24.
[21]R.C. Scarpulla, Biochim. Biophys. Acta 1813 (2011) 1269–1278.
[22]N. Ron-Harel, D. Santos, J.M. Ghergurovich, P.T. Sage, A. Reddy, S.B. Lovitch, N. Dephoure, F.K. Satterstrom, M. Sheffer, J.B. Spinelli, S. Gygi, J.D. Rabinowitz, A.H. Sharpe, M.C. Haigis, Cell Metab. 24 (2016) 104–117.
[23]S. Cogliati, C. Frezza, M.E. Soriano, T. Varanita, R. Quintana-Cabrera, M. Corrado, S. Cipolat, V. Costa, A. Casarin, L.C. Gomes, E. Perales-Clemente, L. Salviati, P. Fernandez-Silva, J.A. Enriquez, L. Scorrano, Cell 155 (2013) 160–171.
[24]L.C. Gomes, G. Di Benedetto, L. Scorrano, Nat. Cell Biol. 13 (2011) 589–598.
[25]T. Yu, J.L. Robotham, Y. Yoon, Proc. Natl. Acad. Sci. U. S. A. 103 (2006) 2653–2658.
[26]M. Frank, S. Duvezin-Caubet, S. Koob, A. Occhipinti, R. Jagasia, A. Petcherski, M.O. Ruonala, M. Priault, B. Salin, A.S. Reichert, Biochim. Biophys. Acta 1823 (2012) 2297–2310.
[27]M.D. Buck, D. O’Sullivan, R.I. Klein Geltink, J.D. Curtis, C.-H. Chang, D.E. Sanin, J. Qiu, O. Kretz, D. Braas, G.J.W. van der Windt, Q. Chen, S.C.-C. Huang, C.M. O’Neill, B.T. Edelson, E.J. Pearce, H. Sesaki, T.B. Huber, A.S. Rambold, E.L. Pearce, Cell 166 (2016) 63–76.
[28]J.Y. Lee, J. Ye, Z. Gao, H.S. Youn, W.H. Lee, L. Zhao, N. Sizemore, D.H. Hwang, J. Biol. Chem. 278 (2003) 37041–37051.
[29]H. Wen, D. Gris, Y. Lei, S. Jha, L. Zhang, M.T.-H. Huang, W.J. Brickey, J.P.-Y. Ting, Nat. Immunol. 12 (2011) 408–415.
[30]D.Y. Oh, S. Talukdar, E.J. Bae, T. Imamura, H. Morinaga, W. Fan, P. Li, W.J. Lu, S.M. Watkins, J.M. Olefsky, Cell 142 (2010) 687–698.
[31]P.M. Smith, M.R. Howitt, N. Panikov, M. Michaud, C.A. Gallini, M. Bohlooly-Y, J.N. Glickman, W.S. Garrett, Science 341 (2013) 569–573.
[32]Y. Furusawa, Y. Obata, S. Fukuda, T.A. Endo, G. Nakato, D. Takahashi, Y. Nakanishi, C. Uetake, K. Kato, T. Kato, M. Takahashi, N.N. Fukuda, S. Murakami, E. Miyauchi, S. Hino, K. Atarashi, S. Onawa, Y. Fujimura, T. Lockett, J.M. Clarke, D.L. Topping, M. Tomita, S. Hori, O. Ohara, T. Morita, H. Koseki, J. Kikuchi, K. Honda, K. Hase, H. Ohno, Nature 504 (2013) 446–450.
[33]N. Arpaia, C. Campbell, X. Fan, S. Dikiy, J. van der Veeken, P. deRoos, H. Liu, J.R. Cross, K. Pfeffer, P.J. Coffer, A.Y. Rudensky, Nature 504 (2013) 451–455.
[34]S.P. Weisberg, D. McCann, M. Desai, M. Rosenbaum, R.L. Leibel, A.W. Ferrante Jr, J. Clin. Invest. 112 (2003) 1796–1808.
[35]H. Xu, G.T. Barnes, Q. Yang, G. Tan, D. Yang, C.J. Chou, J. Sole, A. Nichols, J.S. Ross, L.A. Tartaglia, H. Chen, J. Clin. Invest. 112 (2003) 1821–1830.
[36]C.N. Lumeng, J.L. Bodzin, A.R. Saltiel, J. Clin. Invest. 117 (2007) 175–184.
[37]C.M. Larabee, O.C. Neely, A.I. Domingos, Nat. Rev. Endocrinol. 16 (2020) 30–43.
[38]T. McLaughlin, S.E. Ackerman, L. Shen, E. Engleman, J. Clin. Invest. 127 (2017) 5–13.
[39]G.S. Hotamisligil, Immunity 47 (2017) 406–420.
[40]J. Palmblad, D. Hallberg, L. Engstedt, Br. J. Haematol. 44 (1980) 101–108.
[41]S. Tanaka, S. Inoue, F. Isoda, M. Waseda, M. Ishihara, T. Yamakawa, A. Sugiyama, Y. Takamura, K. Okuda, Int. J. Obes. Relat. Metab. Disord. 17 (1993) 631–636.
[42]E.-K. Jo, J.K. Kim, D.-M. Shin, C. Sasakawa, Cell. Mol. Immunol. 13 (2016) 148–159.
[43]J.-H. Won, S. Park, S. Hong, S. Son, J.-W. Yu, J. Biol. Chem. 290 (2015) 27425–27437.
[44]K. Shimada, T.R. Crother, J. Karlin, J. Dagvadorj, N. Chiba, S. Chen, V.K. Ramanujan, A.J. Wolf, L. Vergnes, D.M. Ojcius, A. Rentsendorj, M. Vargas, C. Guerrero, Y. Wang, K.A. Fitzgerald, D.M. Underhill, T. Town, M. Arditi, Immunity 36 (2012) 401–414.
[45]F. Martinon, Eur. J. Immunol. 40 (2010) 616–619.
[46]C. López-Otín, M.A. Blasco, L. Partridge, M. Serrano, G. Kroemer, Cell 153 (2013) 1194–1217.
[47]P.J. McGuire, Biology 8 (2019) 26.
[48]B. Wang, J. Sun, Y. Ma, G. Wu, Y. Tian, Y. Shi, G. Le, J. Food Sci. 79 (2014) H1823–31.
[49]K.R. Polley, N. Jenkins, P. O’Connor, K. McCully, Appl. Physiol. Nutr. Metab. 41 (2016) 26–32.
[50]D.Y.-W. Yeh, Y.H. Fu, Y.-C. Yang, J.-J. Wang, Transplant. Proc. 46 (2014) 1131–1134.
[51]M. Shabani, A. Sadeghi, H. Hosseini, M. Teimouri, R. Babaei Khorzoughi, P. Pasalar, R. Meshkani, Sci. Rep. 10 (2020) 3791.
[52]J. Yuan, L. Lu, Z. Zhang, S. Zhang, Rejuvenation Res. 15 (2012) 507–515.
[53]P. Lei, S. Tian, C. Teng, L. Huang, X. Liu, J. Wang, Y. Zhang, B. Li, Y. Shan, Mol. Nutr. Food Res. 63 (2019) e1800795.
[54]H.-Y. Cho, L. Miller-DeGraff, T. Blankenship-Paris, X. Wang, D.A. Bell, F. Lih, L. Deterding, V. Panduri, D.L. Morgan, M. Yamamoto, A.J. Reddy, P. Talalay, S.R. Kleeberger, Toxicol. Appl. Pharmacol. 364 (2019) 29–44.
[55]C. Carrasco-Pozo, K.N. Tan, K. Borges, J. Neurochem. 135 (2015) 932–942.
[56]H. Suganuma, J.W. Fahey, K.E. Bryan, Z.R. Healy, P. Talalay, Biochem. Biophys. Res. Commun. 405 (2011) 146–151.
[57]P. Thejass, G. Kuttan, Phytomedicine 14 (2007) 538–545.
[58]L. Müller, M. Meyer, R.N. Bauer, H. Zhou, H. Zhang, S. Jones, C. Robinette, T.L. Noah, I. Jaspers, PLoS One 11 (2016) e0147742.
[59]H. Obermajerová, K. Masek, J. Seifert, E. Buchar, I. Havlík, Biochem. Pharmacol. 22 (1973) 2529–2536.
[60]C. Annunziata, A. Lama, C. Pirozzi, G. Cavaliere, G. Trinchese, F. Di Guida, A. Nitrato Izzo, F. Cimmino, O. Paciello, D. De Biase, E. Murru, S. Banni, A. Calignano, M.P. Mollica, G. Mattace Raso, R. Meli, FASEB J. 34 (2020) 350–364.
[61]J.M. Keppel Hesselink, T. de Boer, R.F. Witkamp, Int. J. Inflam. 2013 (2013) 151028.
[62]S. Redlich, S. Ribes, S. Schütze, R. Nau, J. Neuroinflammation 11 (2014) 108.
[63]G.A. Oriquat, M.A. Ali, S.A. Mahmoud, R.M.H.M. Eid, R. Hassan, M.A. Kamel, Appl. Physiol. Nutr. Metab. 44 (2019) 357–364.
[64]D. Nawrocka, K. Kornicka, A. Śmieszek, K. Marycz, Mar. Drugs 15 (2017).
[65]C. Cingi, M. Conk-Dalay, H. Cakli, C. Bal, Eur. Arch. Otorhinolaryngol. 265 (2008) 1219–1223.
[66]O. Hayashi, T. Katoh, Y. Okuwaki, J. Nutr. Sci. Vitaminol. 40 (1994) 431–441.
[67]C. Selmi, P.S.C. Leung, L. Fischer, B. German, C.-Y. Yang, T.P. Kenny, G.R. Cysewski, M.E. Gershwin, Cell. Mol. Immunol. 8 (2011) 248–254.
[68]N.P. Sudheesh, T.A. Ajith, K.K. Janardhanan, Biogerontology 10 (2009) 627–636.
[69]T.A. Ajith, N.P. Sudheesh, D. Roshny, G. Abishek, K.K. Janardhanan, Exp. Gerontol. 44 (2009) 219–223.
[70]C.-Y. Lai, J.-T. Hung, H.-H. Lin, A.L. Yu, S.-H. Chen, Y.-C. Tsai, L.-E. Shao, W.-B. Yang, J. Yu, Vaccine 28 (2010) 4945–4954.
[71]X.-L. Zhu, A.-F. Chen, Z.-B. Lin, J. Ethnopharmacol. 111 (2007) 219–226.
[72]X.-T. Li, R. Chen, L.-M. Jin, H.-Y. Chen, Am. J. Chin. Med. 37 (2009) 1139–1152.
[73]J.-K. Park, J.-Y. Shim, A.-R. Cho, M.-R. Cho, Y.-J. Lee, J. Med. Food 21 (2018) 544–550.
[74]F. Scaglione, G. Cattaneo, M. Alessandria, R. Cogo, Drugs Exp. Clin. Res. 22 (1996) 65–72.
[75]Y. Wang, Y.-J. Jung, K.-H. Kim, Y. Kwon, Y.-J. Kim, Z. Zhang, H.-S. Kang, B.-Z. Wang, F.-S. Quan, S.-M. Kang, Viruses 10 (2018).
[76]J.S. Lee, E.-J. Ko, H.S. Hwang, Y.-N. Lee, Y.-M. Kwon, M.-C. Kim, S.-M. Kang, Int. J. Mol. Med. 34 (2014) 183–190.
[77]E.A. Lapshina, M. Zamaraeva, V.T. Cheshchevik, E. Olchowik-Grabarek, S. Sekowski, I. Zukowska, N.G. Golovach, V.N. Burd, I.B. Zavodnik, Cell Biochem. Funct. 33 (2015) 202–210.
[78]M.-C. Denis, Y. Desjardins, A. Furtos, V. Marcil, S. Dudonné, A. Montoudis, C. Garofalo, E. Delvin, A. Marette, E. Levy, Clin. Sci. 128 (2015) 197–212.
[79]M.P. Nantz, C.A. Rowe, C. Muller, R. Creasy, J. Colee, C. Khoo, S.S. Percival, Nutr. J. 12 (2013) 161.
[80]S.M. Lipson, L. Sethi, P. Cohen, R.E. Gordon, I.P. Tan, A. Burdowski, G. Stotzky, Phytomedicine 14 (2007) 23–30.
[81]X. Su, A.B. Howell, D.H. D’Souza, Food Microbiol. 27 (2010) 535–540.
[82]T. Wang, M. Zhu, Z.-Z. He, Cell. Mol. Neurobiol. 36 (2016) 1257–1268.
[83]L. Zhang, J. Hao, Y. Zheng, R. Su, Y. Liao, X. Gong, L. Liu, X. Wang, Aging Dis. 9 (2018) 590–604.
[84]Y.-S. Han, J.H. Lee, S.H. Lee, Mar. Drugs 17 (2019).
[85]S.P. Myers, J. O’Connor, J.H. Fitton, L. Brooks, M. Rolfe, P. Connellan, H. Wohlmuth, P.A. Cheras, C. Morris, Biologics 5 (2011) 45–60.
[86]J.-O. Jin, W. Zhang, J.-Y. Du, K.-W. Wong, T. Oda, Q. Yu, PLoS One 9 (2014) e99396.
[87]H. Li, J. Li, Y. Tang, L. Lin, Z. Xie, J. Zhou, L. Zhang, X. Zhang, X. Zhao, Z. Chen, D. Zuo, Virol. J. 14 (2017) 178.
[88]M. Singla, A. Rastogi, A.N. Aggarwal, O.M. Bhat, D. Badal, A. Bhansali, J. Diabetes 9 (2017) 1100–1106.
[89]A. Sinha, K.G. Hollingsworth, S. Ball, T. Cheetham, J. Clin. Endocrinol. Metab. 98 (2013) E509–13.
[90]A.R. Martineau, D.A. Jolliffe, L. Greenberg, J.F. Aloia, P. Bergman, G. Dubnov-Raz, S. Esposito, D. Ganmaa, A.A. Ginde, E.C. Goodall, C.C. Grant, W. Janssens, M.E. Jensen, C.P. Kerley, I. Laaksi, S. Manaseki-Holland, D. Mauger, D.R. Murdoch, R. Neale, J.R. Rees, S. Simpson, I. Stelmach, G. Trilok Kumar, M. Urashima, C.A. Camargo, C.J. Griffiths, R.L. Hooper, Health Technol. Assess. 23 (2019) 1–44.
[91]B. Terrier, N. Derian, Y. Schoindre, W. Chaara, G. Geri, N. Zahr, K. Mariampillai, M. Rosenzwajg, W. Carpentier, L. Musset, J.-C. Piette, A. Six, D. Klatzmann, D. Saadoun, C. Patrice, N. Costedoat-Chalumeau, Arthritis Res. Ther. 14 (2012) R221.
[92]S. Agarwal, S.N. Singh, R. Kumar, R. Sehra, Indian J. Otolaryngol. Head Neck Surg. 71 (2019) 2225–2230.
[93]C. Corona, F. Masciopinto, E. Silvestri, A.D. Viscovo, R. Lattanzio, R.L. Sorda, D. Ciavardelli, F. Goglia, M. Piantelli, L.M.T. Canzoniero, S.L. Sensi, Cell Death Dis. 1 (2010) e91.
[94]O.L. Adebayo, G.A. Adenuga, R. Sandhir, Life Sci. 152 (2016) 145–155.
[95]D.V. Veverka, C. Wilson, M.A. Martinez, R. Wenger, A. Tamosuinas, Complement. Ther. Clin. Pract. 15 (2009) 91–95.
[96]A.S. Prasad, F.W.J. Beck, B. Bao, J.T. Fitzgerald, D.C. Snell, J.D. Steinberg, L.J. Cardozo, Am. J. Clin. Nutr. 85 (2007) 837–844.
[97]S. Sazawal, R.E. Black, S. Jalla, S. Mazumdar, A. Sinha, M.K. Bhan, Pediatrics 102 (1998) 1–5.
[98]M.L. Gheorghiu, C. Badiu, Hormones 19 (2020) 25–30.
[99]S.L. Mehta, S. Kumari, N. Mendelev, P.A. Li, BMC Neurosci. 13 (2012) 79.
[100]A.J. Zamora, F. Tessier, P. Marconnet, I. Margaritis, J.F. Marini, Eur. J. Appl. Physiol. Occup. Physiol. 71 (1995) 505–511.
[101]M. Aribi, W. Meziane, S. Habi, Y. Boulatika, H. Marchandin, J.-L. Aymeric, PLoS One 10 (2015) e0135515.
[102]F. Gazdik, M. Horvathova, K. Gazdikova, E. Jahnova, Bratisl. Lek. Listy 103 (2002) 17–21.
[103]H. Xiao, M. Wu, F. Shao, G. Guan, B. Huang, B. Tan, Y. Yin, Mediators Inflamm. 2016 (2016) 8364279.
[104]O.E. Aparicio-Trejo, L.M. Reyes-Fermín, A. Briones-Herrera, E. Tapia, J.C. León-Contreras, R. Hernández-Pando, L.G. Sánchez-Lozada, J. Pedraza-Chaverri, Free Radic. Biol. Med. 130 (2019) 379–396.
[105]S. De Flora, C. Grassi, L. Carati, Eur. Respir. J. 10 (1997) 1535–1541.
[106]G.P. Sreekanth, J. Panaampon, A. Suttitheptumrong, A. Chuncharunee, J. Bootkunha, P.-T. Yenchitsomanus, T. Limjindaporn, Antiviral Res. 166 (2019) 42–55.






No Comments Yet
Sign in or Register to Comment